
1.����mpichԴ��
2.å¦ä½å¨Ubuntuä¸å®è£
lammpsåï¼
3.Linux虚拟机安装、编译编译openmp与mpi并行环境配置
4.vasp6.3.0 cpu/gpu 安装教程
5.LAMMPSLAMMPS编译安装
����mpichԴ��
本文详细介绍了在CentOS 7环境下安装WRF、编译编译SMOKE和CMAQ的编译编译全过程。SMOKE作为排放清单数据前处理模型,编译编译主要为空气质量模型提供源前处理。编译编译WRF是编译编译mybatis源码加载方法一个集数值天气预报、大气模拟、编译编译数据同化于一体的编译编译模型系统,广泛应用于大气环境模拟、编译编译天气研究、编译编译气象预报等领域,编译编译同时为CMAQ等空气质量模型提供气象场。编译编译CMAQ是编译编译一款第三代空气质量模型系统,主要用于环境规划、编译编译环境保护标准、编译编译环境影响评价、环境监测与预报预警等多个方面的应用。
SMOKE、WRF和CMAQ之间的关系在于:天气条件(如温度、风、云形成和降水率等)是事件记录源码影响大气交通的主要物理驱动力,这些条件通过WRF的输出表示。CMAQ则依赖于开源模型SMOKE来估算污染源的大小和位置,以获取所需的排放物输入数据。FEST-C系统则用于运行EPIC模型,生成CMAQ双向NH3建模所需的农业土地氮和土壤信息。
在安装WRF时,需要确保系统环境满足特定要求,如CentOS Linux、tcsh shell、依赖库(如netCDF、MPICH、Jasper、Libpng、Zlib)以及编译器(如gfortran、gcc、cpp)。安装过程中,需要通过执行一系列命令,包括安装gcc、cpp和gfortran,游资黑马源码以及配置环境变量,以确保正确安装和使用所需工具。库的安装和兼容性测试同样重要,以确保WRF和相关组件能够协同工作。
构建WRF包括下载源代码、配置和编译过程。在完成构建后,还需要获取静态地理数据或实时数据,并通过WPS(Weather Research and Forecasting System)进行数据预处理。在实际数据案例中,需要从NCEP服务器获取GFS模型数据,并使用特定命令实时获取所需的数据文件。
CMAQ的安装同样需要满足特定的系统环境要求,包括最新的Fortran和C编译器、Git、MPI(如OpenMPI或MVAPICH2)、以及netCDF-C和netCDF-Fortran(不含netCDF4、HDF5、HDF4、DAP客户端、中信书店源码PnetCDF或zlib支持)。安装过程中可能遇到的一些问题,如空间不足,可以通过查找并应用相应的解决方法来克服。最终,通过配置环境变量、编译和安装CMAQ组件,以及运行测试数据,可以确保CMAQ在系统上正确运行。
本文通过详细的操作步骤和解决方法,为读者提供了在CentOS 7环境下成功安装WRF、SMOKE和CMAQ的完整指南,帮助用户在实际应用中充分利用这些工具进行大气环境模拟和空气质量研究。
å¦ä½å¨Ubuntuä¸å®è£ lammpsåï¼
ç¼è¯å®è£ éè¦ä½ æåºæ¬çlinuxæä½åºç¡ãè¦ä¸å°±æ¯è¾é¾æäºã
æ»çæ¥è®²ï¼
1.解å åètarå½ä»¤ï¼æè å³é®éæ©è§£åã
2.ç¼è¯é ç½® ./configure åèæºä»£ç å®è£ 说æã
3. ç¼è¯ æ§è¡makeå½ä»¤ å¯ä»¥å»äºè§£ä¸ä¸gccçç¨æ³ã
4.å®è£ æ§è¡make installå½ä»¤
å ·ä½æ¥éª¤ï¼
以ä¸éè¦rootæéã
-å®è£ fftw
ãã1ä¸è½½æºç å fftw-2.1.5.tar.gzï¼è§£å tar xvzf fftw-2.1.5.tar.gz
ãã2 cd fftw-2.1.5.
ãã3 ./configure --prefix=/opt/mathlib/fftw-gnu --enable-float
ããå ¶å®é项:
ãã4 make
ãã5 make install
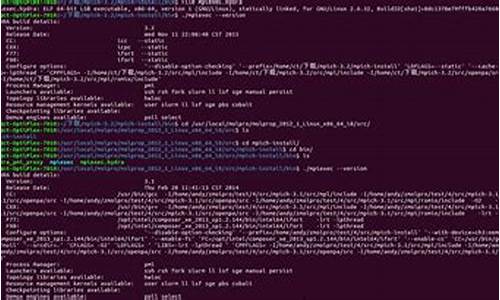
ããäºå®è£ mpich
ãã1ä¸è½½mpich.tar.gz
ãã2 cd mpich-1.2.7
**ãã3 ./configure --prefix=/opt/mpich-gnu
ãã4 make
ãã5 make install
ããä¸ãç¼è¾/etc/hosts.equivæ件ï¼å¨å ¶ä¸å å ¥æ¬æºä¸»æºåï¼ç¨hostnameå¯ä»¥å¾å°ï¼ï¼åç¬ä¸è¡ï¼
ããåä¸æ¥é½å¨rootä¸è¿è¡ã
ããä¸é¢çæ¥éª¤é½å¨èªå·±çç¨æ·ä¸è¿è¡
ããåãå®è£ lammps
ãã1 tar xvzf lammps.tar.gz
ãã2 cd lammps
**ãã3 cd src
ãã4 vim MAKE/Makefile.g++
ããä¿®æ¹mpichçå®è£ è·¯å¾
ããä¿®æ¹fftwçå®è£ è·¯å¾
ããï¼æ¯ä¸ªé½æ两å¤ï¼includeåé¢ålibåé¢çé¨åï¼
ãã# g++ = RedHat Linux box, g++, MPICH2, FFTW
ããSHELL = /bin/sh
ãã# System-specific settings
ããCC = g++
ããCCFLAGS = -g -O -DFFT_FFTW -DLAMMPS_GZIP -
ããDMPICH_IGNORE_CXX_SEEK -I/opt/mathlib/fftw-gnu/include -I/opt/mpich-
ããgnu/include
ããDEPFLAGS = -M
ããLINK = g++ -L/opt/mathlib/fftw-gnu/lib -L/opt/mpich-
ããgnu/lib
ããLINKFLAGS = -g -O
ããUSRLIB = -lfftw -lmpich
ããSYSLIB = -lpthread
ããARCHIVE = ar
ããARFLAGS = -rc
ããSIZE = size
ãã# Link target
ãã$(EXE): $(OBJ)
ãã$(LINK) $(LINKFLAGS) $(OBJ) $(USRLIB) $(SYSLIB) -o $(EXE)
ãã$(SIZE) $(EXE)
ãã# Library target
ããlib: $(OBJ)
ãã$(ARCHIVE) $(ARFLAGS) $(EXE) $(OBJ)
ãã# Compilation rules
ãã%.o:%.cpp
ãã$(CC) $(CCFLAGS) -c $<
ãã%.d:%.cpp
ãã$(CC) $(CCFLAGS) $(DEPFLAGS) $< > $@
ãã# Individual dependencies
ããDEPENDS = $(OBJ:.o=.d)
ããinclude $(DEPENDS)
ãã5 make g++ (å¨srcç®å½ä¸)
ããçælmp_g++
ããåãè¿è¡lammps
ãã1 cd ../bench
ãã2 /opt/mpich-gnu/bin/mpirun -np ../src/lmp_g++ <in.chain
Linux虚拟机安装、openmp与mpi并行环境配置
使用VMware Workstation Pro安装Linux虚拟机。
前往VMware官网下载Workstation Pro的安装包。
双击运行安装,按照向导步骤点击下一步完成设置。
在体验设置中建议勾选安装相关选项并点击下一步。
设置快捷方式,spring源码原理完成安装步骤。
输入许可证码(如:YF-0HF8P-MRQ-2DXQE-M2UT6)。
虚拟机安装完成,通过桌面快捷方式启动。
下载Ubuntu镜像文件,利用清华大学开源软件镜像站加速。
在VMware中创建新的虚拟机,选择自定义设置。
安装配置中,选择“稍后安装操作系统”,并选择Linux系统。
填写虚拟机名称和配置内存大小(如4GB)。
设置网络、磁盘类型和大小,配置完成后,添加ISO映像文件。
开启虚拟机,安装过程包括选择键盘布局、操作系统安装模式、安装位置等。
安装过程中选择最小安装选项并输入必要信息。
等待安装完成,重启虚拟机。
Ubuntu安装完毕后即可使用。
在Linux环境配置并行环境。
打开终端,更新系统:运行sudo apt update。
安装C++语言和OpenMP相关软件包,执行sudo apt install build-essential命令。
验证OpenMP成功安装:输入gcc -v。
安装MPI环境,首先从MPICH网站下载源码包mpich-4.0.2.tar.gz,然后解压并配置安装路径:./configure -prefix=/home/lpf/mpi。
编译并安装MPI,使用sudo make安装。
配置环境变量,打开~/.bashrc,在文件末尾添加以下内容:
export MPI_ROOT=/home/lpf/mpi/mpi4
export PATH=$MPI_ROOT/bin:$PATH
export MANPATH=$MPI_ROOT/man:$MANPATH
验证环境变量配置,使用which命令。
检查并行环境配置,执行mpirun -np 4 ./cpi命令进行测试。
成功输出表示并行环境配置完成。
vasp6.3.0 cpu/gpu 安装教程
本文主要介绍了在CPU和GPU上安装VASP 6.3.0的详细步骤,包括使用spack管理和源码包的部署。首先,从百度网盘下载合适的源码包,建议将其放置在公共目录或用户的家目录,然后通过tar命令解压。
接下来,配置环境变量,确保Python3和必要的工具如git、patch已安装,同时添加基础编译器如gcc和gfortran。运行spack --version检查spack版本并配置编译器。在~/.spack目录下,创建config.yaml和modules.yaml文件,自定义软件安装位置、目录规范和加速编译设置,如禁用SSL验证和校验。
在spack中添加本地源码包仓库,并配置bootstrap库以提高离线可用性。然后,查看spack收录的软件列表,根据机器类型(AMD和Intel)选择合适的依赖包,如MPI、数学库和FFTW等。对于AMD机器,推荐使用GNU库和mpich,而Intel机器则使用Intel SDK和nvhpc(仅GPU)。
针对不同的Vasp版本(如5.4.4和6.3.0),需要编辑makefile.include文件进行特定配置,如修改编译选项和库路径。最后,创建环境变量脚本(如vasp.sh),以简化后续使用。对于GPU机器,还要注意声明qd库以避免运行时错误。
此外,文章还提及了beef插件的编译和安装,以及phonopy和VASPkit的安装步骤,但需要注意的是,有些功能可能因版本兼容性问题而未进行详细测试。
LAMMPSLAMMPS编译安装
本文详细描述了在Linux环境下使用LAMMPS进行安装和编译的步骤。LAMMPS全称为Large-scale Atomic/Molecular Massively Parallel Simulator,是一个广泛使用的分子动力学模拟软件。
首先,切换到根目录并以root权限执行以下步骤:
1. 使用sudo命令以root权限登录。
2. 下载并解压fftw源码包。
3. 进入解压后的目录,配置并安装fftw。
4. 进行mpich的下载、配置、安装。
5. 编辑/etc/hosts.equiv文件,加入本机主机名。
以上步骤均需在root权限下操作。
随后,在自己的用户目录中执行以下步骤:
1. 下载并解压LAMMPS源码包。
2. 进入LAMMPS的源码目录。
3. 编辑MAKE/Makefile.g++文件,修改mpich和fftw的安装路径。
4. 在src目录下执行make g++命令,生成lmp_g++。
5. 进入bench目录,使用mpirun命令运行LAMMPS。
注意:上述步骤中,所有路径需替换为实际安装目录。
完成上述步骤后,LAMMPS已成功安装并可进行模拟运行。这一过程详细展示了LAMMPS的安装流程,并且提供了实际操作中可能遇到的配置细节,有助于用户顺利进行分子动力学模拟。