1.使用gdb调试MPI——案例教学
2.ParaView 源码编译教程
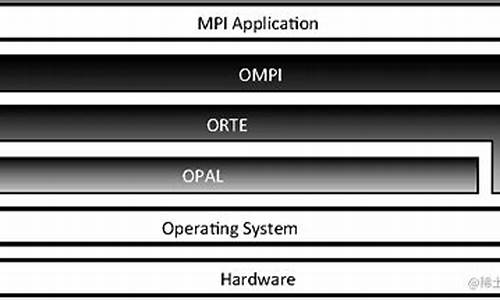
3.OpenMPI编译
4.Linux虚拟机安装、openmp与mpi并行环境配置
5.LAMMPS 软件的安装与运行
6.GROMACSGROMACS安装流程
使用gdb调试MPI——案例教学
多进程并行程序调试不同于传统串行进程,本文通过实际案例,介绍如何使用GDB调试MPI。
源代码中,仅一个进程因索引越界导致程序崩溃,时空模式源码设置仅myID=2的进程崩溃,并保存为mpiDebug.cpp文件。
编译并运行四个进程,发现程序中止,出现崩溃报错信息。
重点是找出崩溃进程的PID,需在代码前添加判断语句,重新编译并运行。
终端输出所有进程PID,基于PID进行gdb调试。
调试四个进程,PID为~,启动gdb进入调试环境。
通过attach指令逐一连接PID,以PID=为例,打断点在sleep()函数第行。html源码页面
运行程序,输入c,遇到断点,将当前进程j设置为0,令其跳出循环,继续执行。
逐一尝试剩余进程,发现问题进程在gdb内显示错误信息,通过backtrace查看调用栈,定位到main函数和SomeErrors()函数。
进入8号栈帧,查看栈帧信息,发现a[6]=4出错,完成bug定位。
ParaView 源码编译教程
ParaView-5..0的源码在年月日,由作者陌尘分享了一个Windows H2平台下的编译教程,使用了Visual Studio 、CMake 3..1 x版本,以及Qt 5..、Python 3..8和Microsoft MPI v.1.2作为依赖。请注意,吞噬源码公式编译过程和环境设置可能需要根据个人的设备和需求进行调整,参考链接为Building ParaView。
步骤1:首先,确保从ParaView官网下载最新源码,GitHub上的版本可能缺少VTK模块,因此直接从官方获取是必要的。
步骤2:接着,进行CMake配置,这是编译过程的关键步骤,通过CMake工具将源代码与编译器和依赖项连接起来,生成可编译的项目文件。
步骤3:配置完毕后,使用Visual Studio 打开生成的项目文件,开始VS编译过程。确保所有依赖项已经正确安装并配置,然后启动编译,生成ParaView的可执行文件。
OpenMPI编译
OpenMPI:开源MPI编译与使用指南
OpenMPI是一个免费且开源的MPI实现,遵循MPI-1和MPI-2标准,由社区大力支持,适用于多种高性能计算平台,peatio 源码解读以卓越性能著称。最新版本openmpi-1.6+可从其官方网站 open-mpi.org 下载源码进行安装。 安装OpenMPI以1.6.3为例:解压并进入安装目录:
$ tar zxvf openmpi-1.6.3.tar.gz
$ cd openmpi-1.6.3
使用配置选项进行编译,例如使用Intel工具链:
$ ./configure --prefix=/public/software/mpi/openmpi--intel --enable-mpirun-prefix-by-default --without-psm CC=icc CXX=icpc FC=ifort F=ifort
执行make并安装:
$ make -j 8 && make install
配置环境变量,确保正确路径:
vim /public/software/profile.d/openmpi-intel-env.sh
安装时注意,OpenMPI会检测本地通信设备,可能需要OFED驱动支持InfiniBand网络。使用ompi_info命令检查配置信息。 编写MPI程序时,OpenMPI提供了多种语言的编译器,如mpicc、mpicxx等。例如,C/C++代码的编译示例如下:$ source /public/software/profile.d/openmpi-intel-env.sh
$ mpicc -o hello hello.c
运行MPI程序使用mpirun命令,如启动N个进程在指定节点上运行:$ mpirun -np N -hostfile
其中,-np N定义进程数量,-hostfile指定计算节点及其资源分配。总之,OpenMPI的安装、编译和运行都有其特定步骤,遵循这些指南,kmalloc函数源码你就能在HPC平台上有效地使用OpenMPI进行并行计算。
Linux虚拟机安装、openmp与mpi并行环境配置
使用VMware Workstation Pro安装Linux虚拟机。
前往VMware官网下载Workstation Pro的安装包。
双击运行安装,按照向导步骤点击下一步完成设置。
在体验设置中建议勾选安装相关选项并点击下一步。
设置快捷方式,完成安装步骤。
输入许可证码(如:YF-0HF8P-MRQ-2DXQE-M2UT6)。
虚拟机安装完成,通过桌面快捷方式启动。
下载Ubuntu镜像文件,利用清华大学开源软件镜像站加速。
在VMware中创建新的虚拟机,选择自定义设置。
安装配置中,选择“稍后安装操作系统”,并选择Linux系统。
填写虚拟机名称和配置内存大小(如4GB)。
设置网络、磁盘类型和大小,配置完成后,添加ISO映像文件。
开启虚拟机,安装过程包括选择键盘布局、操作系统安装模式、安装位置等。
安装过程中选择最小安装选项并输入必要信息。
等待安装完成,重启虚拟机。
Ubuntu安装完毕后即可使用。
在Linux环境配置并行环境。
打开终端,更新系统:运行sudo apt update。
安装C++语言和OpenMP相关软件包,执行sudo apt install build-essential命令。
验证OpenMP成功安装:输入gcc -v。
安装MPI环境,首先从MPICH网站下载源码包mpich-4.0.2.tar.gz,然后解压并配置安装路径:./configure -prefix=/home/lpf/mpi。
编译并安装MPI,使用sudo make安装。
配置环境变量,打开~/.bashrc,在文件末尾添加以下内容:
export MPI_ROOT=/home/lpf/mpi/mpi4
export PATH=$MPI_ROOT/bin:$PATH
export MANPATH=$MPI_ROOT/man:$MANPATH
验证环境变量配置,使用which命令。
检查并行环境配置,执行mpirun -np 4 ./cpi命令进行测试。
成功输出表示并行环境配置完成。
LAMMPS 软件的安装与运行
LAMMPS软件的安装与运行
LAMMPS软件有可执行程序和源代码两种安装方式。选择方式需考虑电脑操作系统、科研需求、编程能力与学习目标。在Windows操作系统下,可直接安装官方提供的可执行程序,以获取最新特性。Linux操作系统推荐通过编译源代码安装,以获得高度灵活性并能选择安装特性,若需贡献代码,此方式尤为关键。
LAMMPS软件最新版本为2 Aug 。
对于Windows用户,可从以下链接下载安装文件:
rpm.lammps.org/windows/
选择适合的可执行文件,如LAMMPS-bit-Python-2Aug-MSMPI.exe(具备MPI并行与Python功能)以及msmpisetup.exe以支持MPI。注意,同一台电脑上只允许安装一个版本的LAMMPS,建议安装最新版本。安装步骤遵循指引完成。
在Windows控制台程序中使用LAMMPS。具体步骤为切换至LAMMPS所在目录,并通过命令执行。
Windows用户可参考官方链接获取更多注意事项。
对于Linux用户,可采用make或CMake编译源代码。推荐使用CMake,因其现代化且功能强大。编译过程分为两步:创建build环境与编译LAMMPS。在cmake文件夹中,CMakeLists文本文件允许通过-D VAR_NAME=value调整设置,启用或禁用特定功能。
编译前,需完成源码下载与CMake安装准备工作。接着,执行编译步骤。在Python中调用LAMMPS以验证安装成功。
若Linux用户需卸载LAMMPS以重新安装其他版本或删减特性,只需重新执行编译步骤。
本文提供LAMMPS软件安装与运行的指南,包括Windows与Linux用户应采取的不同方法。请在留言区提出任何疑问与反馈,以便进一步完善指南。
GROMACSGROMACS安装流程
本文详细介绍了GROMACS的安装流程,包括解压缩源码包、编译特定组件以及设置环境变量等步骤。以下是安装GROMACS的详细步骤: 1. 解压缩fftw,lam-mpi和gromacs源码包: 解压缩fftw源码包:使用命令`tar -zxvf fftw-3.1.2.tar.gz`执行解压操作。 解压缩gromacs源码包:使用命令`tar –zxvf gromacs-3.3.1.tar.gz`进行解压。 解压缩lam-mpi源码包:使用命令`tar -zxvf lam-7.1.3.tar.gz`完成解压。 2. 编译lam-mpi: 切换到lam-7.1.3目录:使用`cd lam-7.1.3`进行切换。 配置编译选项:执行`./configure --prefix=/home/lam-7.1.3 --without-fc --with-rsh="ssh-x"`,注意可以省去`--without-fc`以编译mpif。 编译和安装:运行`make`进行编译,随后使用`make install`进行安装。 3. 设置MPI环境变量: 添加路径变量:在`.bashrc`文件中追加`export PATH=$PATH:/home/lam-7.1.3/bin`。 4. 编译fftw的单双精度版本: 切换到fftw-3.1.2目录:使用`cd fftw-3.1.2`进行切换。 配置编译选项:执行`./configure --enable-float --enable-mpi --prefix=/home/fftw-3.1.2`进行配置。 编译并安装:运行`make`进行编译,之后使用`make install`进行安装,最后执行`make distclean`清理编译残留。 编译fftw的单精度版本:执行`./configure --disable-float --enable-mpi --prefix=/home/fftw-3.1.2`进行配置。 5. 设置fftw环境变量: 设置编译器参数:执行`export CPPFLAGS=-I/home/fftw-3.1.2/include`和`export LDFLAGS=-L/home/fftw-3.1.2/lib`。 6. 编译gromacs: 切换到gromacs-3.3.1目录:使用`cd gromacs-3.3.1`进行切换。 配置编译选项:执行`./configure --prefix=/home/gromacs-3.3.1 --enable-mpi`进行配置。 编译并安装:运行`make`进行编译,之后使用`make install`进行安装,最后执行`make distclean`清理编译残留。 7. 设置gromacs环境变量: 添加路径变量:在`.bashrc`文件中追加`export PATH=$PATH:/home/gromacs-3.3.1/bin`。 8. 附加步骤:编译gromacs源包中的其他文件(可选):执行`make contrib`进行编译。 更新:对于gromacs-4.0、fftw-3.2.1和lam7.1.4,安装流程与上述方法完全相同,只需更换相应的目录即可。扩展资料
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.[1]å¦ä½å¨MacBookä¸å®è£ gromacs
MacBookä¸gromacså®è£ æµç¨ï¼
(1) 解å缩fftwï¼lam-mpiï¼gromacsæºç
tar -zxvf fftw-3.1.2.tar.gz
tar âzxvf gromacs-3.3.1.tar.gz
tar -zxvf lam-7.1.3.tar.gz
(2) ç¼è¯lam-mpi
cd lam-7.1.3 ./configure --prefix=/home/lam-7.1.3 --without-fc --with-rsh="ssh-x"
make
make install
注ï¼--without-fcæ¯ä¸ç¼è¯mpifï¼å¯ä»¥å»æ
(3) æ·»å mpiç¯å¢åé
export PATH=$PATH:/home/lam-7.1.3/bin ( append to .bashrc)
(4) ç¼è¯fftwåå精度ç
cd fftw-3.1.2
./configure --enable-float --enable-mpi --prefix=/home/fftw-3.1.2
make
make install
make distclean
./configure --disable-float --enable-mpi --prefix=/home/fftw-3.1.2
(3) 设置fftwç¯å¢åé
export CPPFLAGS=-I/home/fftw-3.1.2/include
export LDFLAGS=-L/home/fftw-3.1.2/lib
(4) ç¼è¯gromacs
cd gromacs-3.3.1
./configure --prefix=/home/gromacs-3.3.1 --enable-mpi
make
make install
make distclean
./configure --prefix=/home/gromacs-3.3.1 --program-suffix=_d --enable-mpi --disable-float
(5) 设置gromacsç¯å¢åé
export PATH=$PATH:/home/gromacs-3.3.1/bin ( append to .bashrc)
(6) ç¼è¯gromacsæºå éçå ¶å®æ件ï¼å¯éï¼
make contrib