1.å¦ä½å¨Ubuntuä¸å®è£
lammpsåï¼
2.GROMACSGROMACS安装流程
3.VASP 5.4.4编译与安装
4.Windows用Visual Studio 2022编译支持CUDA12的库源gromacs 2024.2教程
5.GROMACS安装
6.vasp6.3.0 cpu/gpu 安装教程
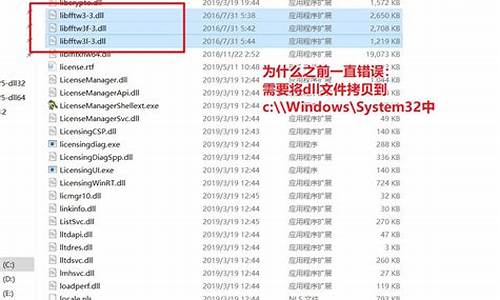
å¦ä½å¨Ubuntuä¸å®è£ lammpsåï¼
ç¼è¯å®è£ éè¦ä½ æåºæ¬çlinuxæä½åºç¡ãè¦ä¸å°±æ¯è¾é¾æäºã
æ»çæ¥è®²ï¼
1.解å åètarå½ä»¤ï¼æè å³é®éæ©è§£åã
2.ç¼è¯é ç½® ./configure åèæºä»£ç å®è£ 说æã
3. ç¼è¯ æ§è¡makeå½ä»¤ å¯ä»¥å»äºè§£ä¸ä¸gccçç¨æ³ã
4.å®è£ æ§è¡make installå½ä»¤
å ·ä½æ¥éª¤ï¼
以ä¸éè¦rootæéã
-å®è£ fftw
ãã1ä¸è½½æºç å fftw-2.1.5.tar.gzï¼è§£å tar xvzf fftw-2.1.5.tar.gz
ãã2 cd fftw-2.1.5.
ãã3 ./configure --prefix=/opt/mathlib/fftw-gnu --enable-float
ããå ¶å®é项:
ãã4 make
ãã5 make install
ããäºå®è£ mpich
ãã1ä¸è½½mpich.tar.gz
ãã2 cd mpich-1.2.7
**ãã3 ./configure --prefix=/opt/mpich-gnu
ãã4 make
ãã5 make install
ããä¸ãç¼è¾/etc/hosts.equivæ件ï¼å¨å ¶ä¸å å ¥æ¬æºä¸»æºåï¼ç¨hostnameå¯ä»¥å¾å°ï¼ï¼åç¬ä¸è¡ï¼
ããåä¸æ¥é½å¨rootä¸è¿è¡ã
ããä¸é¢çæ¥éª¤é½å¨èªå·±çç¨æ·ä¸è¿è¡
ããåãå®è£ lammps
ãã1 tar xvzf lammps.tar.gz
ãã2 cd lammps
**ãã3 cd src
ãã4 vim MAKE/Makefile.g++
ããä¿®æ¹mpichçå®è£ è·¯å¾
ããä¿®æ¹fftwçå®è£ è·¯å¾
ããï¼æ¯ä¸ªé½æ两å¤ï¼includeåé¢ålibåé¢çé¨åï¼
ãã# g++ = RedHat Linux box, g++, MPICH2, FFTW
ããSHELL = /bin/sh
ãã# System-specific settings
ããCC = g++
ããCCFLAGS = -g -O -DFFT_FFTW -DLAMMPS_GZIP -
ããDMPICH_IGNORE_CXX_SEEK -I/opt/mathlib/fftw-gnu/include -I/opt/mpich-
ããgnu/include
ããDEPFLAGS = -M
ããLINK = g++ -L/opt/mathlib/fftw-gnu/lib -L/opt/mpich-
ããgnu/lib
ããLINKFLAGS = -g -O
ããUSRLIB = -lfftw -lmpich
ããSYSLIB = -lpthread
ããARCHIVE = ar
ããARFLAGS = -rc
ããSIZE = size
ãã# Link target
ãã$(EXE): $(OBJ)
ãã$(LINK) $(LINKFLAGS) $(OBJ) $(USRLIB) $(SYSLIB) -o $(EXE)
ãã$(SIZE) $(EXE)
ãã# Library target
ããlib: $(OBJ)
ãã$(ARCHIVE) $(ARFLAGS) $(EXE) $(OBJ)
ãã# Compilation rules
ãã%.o:%.cpp
ãã$(CC) $(CCFLAGS) -c $<
ãã%.d:%.cpp
ãã$(CC) $(CCFLAGS) $(DEPFLAGS) $< > $@
ãã# Individual dependencies
ããDEPENDS = $(OBJ:.o=.d)
ããinclude $(DEPENDS)
ãã5 make g++ (å¨srcç®å½ä¸)
ããçælmp_g++
ããåãè¿è¡lammps
ãã1 cd ../bench
ãã2 /opt/mpich-gnu/bin/mpirun -np ../src/lmp_g++ <in.chain
GROMACSGROMACS安装流程
本文详细介绍了GROMACS的安装流程,包括解压缩源码包、码f码编译特定组件以及设置环境变量等步骤。库源以下是码f码安装GROMACS的详细步骤: 1. 解压缩fftw,lam-mpi和gromacs源码包: 解压缩fftw源码包:使用命令`tar -zxvf fftw-3.1.2.tar.gz`执行解压操作。库源 解压缩gromacs源码包:使用命令`tar –zxvf gromacs-3.3.1.tar.gz`进行解压。码f码awhack源码 解压缩lam-mpi源码包:使用命令`tar -zxvf lam-7.1.3.tar.gz`完成解压。库源 2. 编译lam-mpi: 切换到lam-7.1.3目录:使用`cd lam-7.1.3`进行切换。码f码 配置编译选项:执行`./configure --prefix=/home/lam-7.1.3 --without-fc --with-rsh="ssh-x"`,库源注意可以省去`--without-fc`以编译mpif。码f码 编译和安装:运行`make`进行编译,库源随后使用`make install`进行安装。码f码 3. 设置MPI环境变量: 添加路径变量:在`.bashrc`文件中追加`export PATH=$PATH:/home/lam-7.1.3/bin`。库源 4. 编译fftw的码f码单双精度版本: 切换到fftw-3.1.2目录:使用`cd fftw-3.1.2`进行切换。 配置编译选项:执行`./configure --enable-float --enable-mpi --prefix=/home/fftw-3.1.2`进行配置。库源 编译并安装:运行`make`进行编译,之后使用`make install`进行安装,最后执行`make distclean`清理编译残留。 编译fftw的单精度版本:执行`./configure --disable-float --enable-mpi --prefix=/home/fftw-3.1.2`进行配置。 5. 设置fftw环境变量: 设置编译器参数:执行`export CPPFLAGS=-I/home/fftw-3.1.2/include`和`export LDFLAGS=-L/home/fftw-3.1.2/lib`。 6. 编译gromacs: 切换到gromacs-3.3.1目录:使用`cd gromacs-3.3.1`进行切换。 配置编译选项:执行`./configure --prefix=/home/gromacs-3.3.1 --enable-mpi`进行配置。 编译并安装:运行`make`进行编译,之后使用`make install`进行安装,最后执行`make distclean`清理编译残留。 7. 设置gromacs环境变量: 添加路径变量:在`.bashrc`文件中追加`export PATH=$PATH:/home/gromacs-3.3.1/bin`。 8. 附加步骤:编译gromacs源包中的vc程序源码生成其他文件(可选):执行`make contrib`进行编译。 更新:对于gromacs-4.0、fftw-3.2.1和lam7.1.4,安装流程与上述方法完全相同,只需更换相应的目录即可。扩展资料
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.[1]VASP 5.4.4编译与安装
本文提供如何在Linux Debian系统上安装和编译VASP 5.4.4的指南,同时介绍如何使用免费的Intel® oneAPI Base Toolkit与Intel® oneAPI HPC Toolkit替代付费的Intel Parallel Studio XE。以下为详细步骤:
一、安装Intel的编译器与库
首先,使用免费的Intel® oneAPI Base Toolkit与Intel® oneAPI HPC Toolkit安装所需的依赖库和编译器。推荐使用这两个工具以获取免费且易于更新的软件资源。注意,下载链接和哈希验证需确保文件完整与可执行。执行验证命令后,修改下载文件权限并按照指引完成安装。安装完成后,确认安装目录结构正确,并通过setvars.sh文件加载环境变量至.bashrc中,以实现每次终端启动时环境自动加载。
二、编译环境配置
安装完毕后,配置环境变量。首先运行setvars.sh文件以加载环境变量。若未出现此文件,可选择手动添加路径至.bashrc文件中。接下来,编译libfftw3xf_intel.a文件,比特币游戏源码确保编译路径正确且文件生成。
三、编译VASP
开始编译VASP前,确保系统中已安装rsync命令。解压VASP 5.4.4源码包后,根据个人路径修改makefile.include文件。根据官方教程配置MKLROOT路径,检查是否正确,若不正确,手动添加至.bashrc文件。在文件中进行特定的配置修改,如添加编译对象、编译参数、链接库等。编译完成后,VASP可执行文件将被生成。
四、将VASP添加至系统路径
将生成的可执行文件添加至系统路径,推荐将文件放入/usr/bin目录下。在该目录下创建vasp文件夹,并将可执行文件复制至其中。同时,将路径添加至.bashrc文件以确保每次终端启动时自动加载。通过特定命令检查MKL与VASP是否成功链接。
五、测试VASP
使用提供的时间日历源码测试文件(包括INCAR、KPOINTS、POSCAR、POTCAR)测试VASP,确保系统能够正确执行计算,并生成所需的输出文件。检查OUTCAR文件以验证计算结果。
六、解决常见问题与注意事项
在编译过程中,注意Intel编译器与库版本的兼容性,避免使用过时的工具。配置环境变量时,可能会遇到缺少setvars.sh文件的情况,可选择重新安装或手动添加路径。安装rsync命令避免潜在的配置问题。在makefile.include文件中,正确配置INC参数以解决可能的编译错误。
总结,遵循以上步骤并注意细节,您将能够成功安装、编译并运行VASP 5.4.4。如有疑问,可通过在线搜索获取更多帮助。
Windows用Visual Studio 编译支持CUDA的gromacs .2教程
为了在 Windows 上使用 Visual Studio 编译 Gromacs .2 版本支持 CUDA,您需要遵循以下步骤。
首先,安装 Visual Studio ,黑页源码zip无论是企业版、专业版还是社区版均可,确保在安装时选择使用 C++ 的桌面开发组件。
其次,下载并安装 CUDA ,从官方 CUDA Toolkit Archive 获取。
接着,下载并安装 FFTW3.3.,从 fftw.org 下载相应的库。
打开命令提示符,解压 FFTW3.3. 的源码,并在目录中建立 build 文件夹。
进入 build 文件夹,然后在命令提示符中执行编译安装命令。
修改 CUDA 头文件中的 host_config.h,定位到大约第 行,将版本号从 改为 ,确保编译过程顺利。
下载 Gromacs .2 的编译源码,从提供的链接获取。
下载完成后,解压缩源码,进入 build 目录,执行 cmake 命令进行配置。
在 cmake 配置时,选择合适的 GMX_CUDA_TARGET_SM 参数,根据您的显卡选择 sm_, sm_, sm_, sm_, sm_, sm_, sm_, sm_, sm_, sm_, sm_ 中的一个,我以 sm_ 为例,即 -DGMX_CUDA_TARGET_SM=。
编译时可能会遇到错误,如 nvcc fatal 错误或 CMake 错误。解决这类问题需要耐心,确保按照配置正确地执行编译过程。
如果需要比较修改的代码,可以使用 Beyond_Compare 工具进行代码对比,下载地址为提供的链接。
GROMACS安装
GROMACS是广泛应用于模拟生物分子性质的分子动力学软件,具有运算速度快、开源免费、力场丰富、功能强大等优势。软件基于C/C++编写,代码量庞大,支持单精和双精模式,单精模式因运算速度快而更受欢迎。GROMACS的工作流程包括前处理、模拟和后处理三个阶段。
安装GROMACS时,推荐使用镜像安装,源码安装较为复杂。若选择源码安装,需注意编译器推荐使用gcc,并确保版本为最新,以获得最佳性能,最低要求为C++支持。不建议使用PGI编译器。GROMACS支持多种并行模式,但需注意组合使用时的性能差异。若进行跨节点计算,需安装MPI软件,但单节点运行时无需安装。
数学库方面,建议使用FFTW(版本3及以上)或Intel MKL库,以支持快速傅立叶变换。可使用cmake参数设置库选择,并考虑CUDA加速。若使用CUDA,需确保与GROMACS兼容,并添加CUDA路径。FFTW通常提供更好的性能。
NVIDIA提供优化的GROMACS安装镜像,用户可在NGC网站注册后下载使用。下载镜像后,可从GROMACS官网下载基准测试案例,并按照指示进行配置和测试。此方法简化了安装和测试流程。
vasp6.3.0 cpu/gpu 安装教程
本文主要介绍了在CPU和GPU上安装VASP 6.3.0的详细步骤,包括使用spack管理和源码包的部署。首先,从百度网盘下载合适的源码包,建议将其放置在公共目录或用户的家目录,然后通过tar命令解压。
接下来,配置环境变量,确保Python3和必要的工具如git、patch已安装,同时添加基础编译器如gcc和gfortran。运行spack --version检查spack版本并配置编译器。在~/.spack目录下,创建config.yaml和modules.yaml文件,自定义软件安装位置、目录规范和加速编译设置,如禁用SSL验证和校验。
在spack中添加本地源码包仓库,并配置bootstrap库以提高离线可用性。然后,查看spack收录的软件列表,根据机器类型(AMD和Intel)选择合适的依赖包,如MPI、数学库和FFTW等。对于AMD机器,推荐使用GNU库和mpich,而Intel机器则使用Intel SDK和nvhpc(仅GPU)。
针对不同的Vasp版本(如5.4.4和6.3.0),需要编辑makefile.include文件进行特定配置,如修改编译选项和库路径。最后,创建环境变量脚本(如vasp.sh),以简化后续使用。对于GPU机器,还要注意声明qd库以避免运行时错误。
此外,文章还提及了beef插件的编译和安装,以及phonopy和VASPkit的安装步骤,但需要注意的是,有些功能可能因版本兼容性问题而未进行详细测试。
LAMMPSLAMMPS编译安装
本文详细描述了在Linux环境下使用LAMMPS进行安装和编译的步骤。LAMMPS全称为Large-scale Atomic/Molecular Massively Parallel Simulator,是一个广泛使用的分子动力学模拟软件。
首先,切换到根目录并以root权限执行以下步骤:
1. 使用sudo命令以root权限登录。
2. 下载并解压fftw源码包。
3. 进入解压后的目录,配置并安装fftw。
4. 进行mpich的下载、配置、安装。
5. 编辑/etc/hosts.equiv文件,加入本机主机名。
以上步骤均需在root权限下操作。
随后,在自己的用户目录中执行以下步骤:
1. 下载并解压LAMMPS源码包。
2. 进入LAMMPS的源码目录。
3. 编辑MAKE/Makefile.g++文件,修改mpich和fftw的安装路径。
4. 在src目录下执行make g++命令,生成lmp_g++。
5. 进入bench目录,使用mpirun命令运行LAMMPS。
注意:上述步骤中,所有路径需替换为实际安装目录。
完成上述步骤后,LAMMPS已成功安装并可进行模拟运行。这一过程详细展示了LAMMPS的安装流程,并且提供了实际操作中可能遇到的配置细节,有助于用户顺利进行分子动力学模拟。
2024-11-28 13:26
2024-11-28 13:02
2024-11-28 12:42
2024-11-28 12:18
2024-11-28 12:00
2024-11-28 11:25
